Le site des ophtalmologistes de France
Espace Encyclopédie
Encyclopédie de la vue

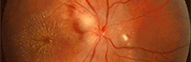




Atrophie Optique dominante Labo UMR 5088 et INSERM U583
Présentation

Photo 2 : Groupe de recherche dirigé par Pascale Belenguer (UMR5088, Dir Bernard Ducommun) à Toulouse. De gauche à droite : au 1er rang : Pascale Belenguer (enseignant-chercheur, Université Paul Sabatier), Emmanuelle Guillou (étudiante en thèse, UPS), Marlène Daloyau (Technicienne, CNRS), au 2ème rang : Guy Lenaers (chercheur, CNRS), Aurélien Olichon (étudiant en thèse, UPS), Laetitia Pelloquin (enseignant-chercheur, UPS), Laurent Emorine (chercheur, CNRS), Céline Bousquet (étudiante en maîtrise, UPS), Maher Costantine (étudiant en DEA, UPS).
Cliquer pour agrandir
Click to enlarge
En collaboration avec l’équipe du Dr Christian Hamel à Montpellier (INSERM U583), nous avons montré que des mutations sur le gène codant pour OPA1 sont responsables de l’atrophie optique dominante de type 1 ou maladie de Kjer (OPA1, MIM 165500).
Cette pathologie héréditaire à transmission dominante est la conséquence d’une dégénérescence des neurones ganglionnaires de la rétine dont les axones forment le nerf optique (photo 5). Elle mène à une perturbation de la vision des couleurs et à une détérioration progressive de l'acuité visuelle pouvant aller jusqu’à la cécité.
Avec une fréquence de l'ordre de 1 pour 50000, la maladie de Kjer est, avec le syndrome de Leber ou LHON (MIM535000), une des formes majeures d’atrophie optique héréditaire. Il est intéressant de noter que le syndrome de Leber est également une pathologie mitochondriale mais liée à des mutations du génome mitochondrial. Ces deux pathologies, contrairement à la plupart des maladies mitochondriales, induisant (Kearns-Sayre voir MIM530000, NARP voir MIM 551500) ou non des syndromes visuels, semblent ne toucher qu’un seul tissu, l’œil, en affectant dans les deux cas les mêmes cellules de la rétine.
Grâce au soutien de Rétina nous menons des travaux afin de rechercher les fondements moléculaires de la maladie de Kjer en explorant les conséquences des mutations de la protéine OPA1 sur la forme du réseau des mitochondries, leur fonctionnement et la stabilité de leur génome, ainsi que leurs effets sur la dégénérescence des neurones ganglionnaires de la rétine.
Nous espérons que la découverte du gène responsable de l’atrophie optique dominante de type 1 conduira à une amélioration de la prise en charge clinique des patients atteints (incidence réelle de la maladie, gravité, mise en place d’un diagnostic simplifié…) et qu’à plus long terme la description de la fonction exacte de OPA1 et des mécanismes physiopathologiques conduisant à la neuropathie optique de Kjer permettra d’envisager d’éventuelles perspectives thérapeutiques.
Les mitochondries
Les mitochondries, siège de la respiration cellulaire, fournissent l’énergie vitale des cellules. Leur dysfonctionnement est à l’origine de nombreuses pathologies dont les symptômes sont variés (cardiaques, hépatiques, neurologiques, auditifs, oculaires…), et souvent combinés.
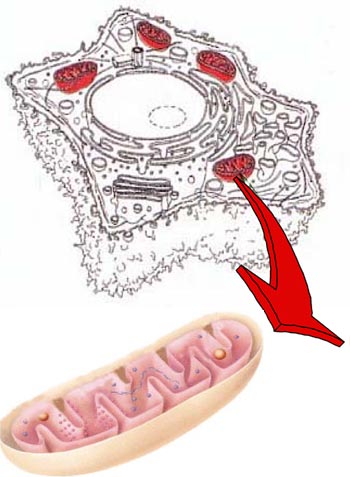
Photo 1 : Dans la cellule les mitochondries (en rouge) sont des organites à double membrane : la membrane externe (en beige) et la membrane interne (en rose). Cette dernière forme des repliements, les crêtes, qui portent les machineries responsables de la respiration et de la production d’énergie cellulaire (boules roses). L’espace entre les deux membranes est appelé espace intermembranaire. La matrice, délimitée par la membrane interne, contient le génome mitochondrial (hélice violette), les ribosomes qui synthétisent les protéines codées par le génome mitochondrial (boules violettes) et d’autres composants impliqués dans le métabolisme de la cellule. Dessins adaptés de Lodisch et al. et Cooper G.M.
Un mauvais fonctionnement des mitochondries peut résulter de mutations qui touchent le génome des mitochondries, ou celui du noyau qui est à l’origine de la synthèse de la grande majorité des protéines mitochondriales. Les mitochondries sont des organites à double membrane (photo 1), qui forment dans la cellule un réseau dont la morphologie est contrôlée par des processus de fission ou de fusion.
Bien que ces mécanismes restent largement à élucider, des protéines nommées Dnm1/DRP1 et Fzo1/Mfn1-2, sont toutefois connues pour intervenir dans la fission et la fusion de la membrane mitochondriale externe. Notre équipe de recherche (photo 2), a découvert une protéine nommée Msp1, codée par le génome nucléaire et localisée dans la mitochondrie de la levure S. pombe au niveau de la membrane interne. La disparition de Msp1 entraîne une altération de la forme du réseau des mitochondries (photo 3), une diminution de la respiration des cellules, ainsi que la perte du génome mitochondrial qui à terme provoque la mort des cellules. Cette protéine jouerait donc un rôle clé dans la dynamique de la membrane interne et le maintien de l’ADN mitochondrial. Notre équipe a découvert OPA1, l’équivalent humain de Msp1. La disparition de cette protéine mitochondriale de la membrane interne perturbe la forme du réseau des mitochondries (photo 4), et provoque la mort des cellules par apoptose.
Photo 3 : Levures S. pombe visualisées en microscopie à contraste de phase interférentiel et à fluorescence. Les mitochondries d’une levure, exprimant la protéine Msp1 normale (gauche) ou mutée (droite), apparaissent en vert. Le réseau mitochondrial continu (gauche) est rompu quand la protéine Msp1 mutante est exprimée (droite). Clichés fournis par Emmanuelle Guillou, UMR5088 Toulouse.
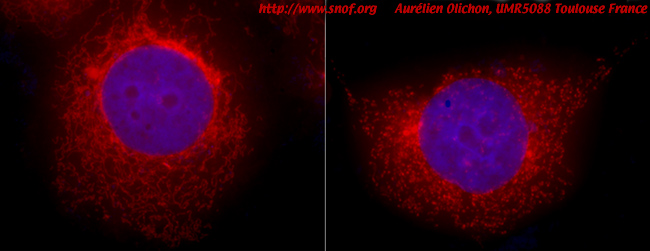
Photo 4 : Cellules humaines visualisées en microscopie à fluoresence. Le réseau mitochondrial de cellules, exprimant (haut) ou n’exprimant pas (bas) la protéine OPA1, apparaît en rouge et le noyau en violet. En présence de la protéine OPA1, les mitochondries forment un réseau filamenteux, en son absence les mitochondries apparaissent sous forme de petits points. Clichés fournis par Aurélien Olichon, UMR5088 Toulouse.
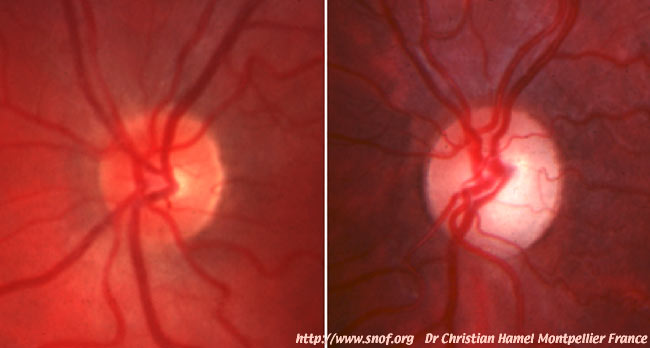
Photo 5 : Fond d’oeil d’un individu sain (gauche) et d’un patient atteint de la maladie de Kjer (droite) porteur d’une mutation du gène codant pour la protéine OPA1. Chez le patient le disque optique atrophique (point d’émergence du nerf optique au fond d’oeil) apparaît blanc pale et non jaune orangé comme chez l’individu sain. Clichés fournis par le Dr C. Hamel, U583 INSERM Montpellier

![]()
Suivez nous sur...
![]()
SNOF
10 rue Schweighaeuser
CS 40028
67080 STRASBOURG Cedex
Tél. 03 88 35 01 09
Fax. 03 88 25 51 90