Dystrophies héréditaires de la rétine
Rétinopathie pigmentaire
Retinitis pigmentosa (RP)
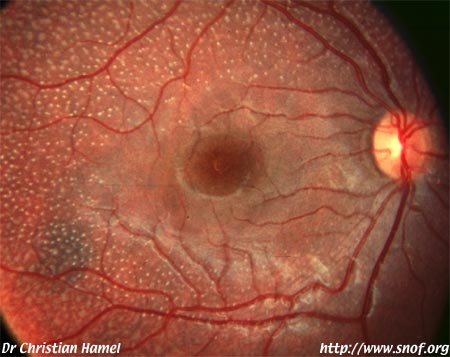
Mutation RDH5 flavimaculée
Nous remercions, pour sa très belle iconographie, le Docteur Christian Hamel Directeur de Recherches Responsable de l'équipe génétique Inserm U. 254 Montpellier France.
L'étude des rétinopathies pigmentaires est difficile, surtout dans son approche génétique. Nous nous proposons donc d'améliorer progressivement ce chapitre.
Plan
- Introduction
- Historique des rétinopathies pigmentaires
- Clinique
- Electrophysiologie
- Evolution
- Notions de génétique
- Rhodopsine
- Biologie moléculaire
- Classement des dystrophies héréditaires de la rétine
- Classification des rétinopathies pigmentaires
- Mutations
- Consultations de génétique ophtalmologique
- Bibliographie
1) Introduction
Les rétinopathies pigmentaires représentent un groupe de maladies génétiques, caractérisées par la perte progressive des photorécepteurs et le dysfonctionnement de l'épithélium pigmentaire, associés à des dépôts pigmentaires visibles au fond d'oeil.
Elles aboutissent souvent à la cécité, mais leur grande hétérogénéité phénotypique et génotypique rend leur étude complexe et le conseil génétique difficile pour les familles.
Leur prévalence est d'environ 1/4000 naissances. On évalue à 30.000 le nombre de patients atteints de rétinopathies pigmentaires en France.
Leur étude permet de mieux appréhender les dysfonctionnements génétiques qui sont à l'origine de ces troubles visuels. Il existe des familles entières qui sont atteintes, parfois de façon plus ou moins grave. Dans d'autres cas, ce sont des individus isolés qui sont porteurs de ces maladies. La plupart de ces maladies sont transmises sur un mode mendéleien, quelques unes sont transmises par des mutations mitochondriales ou par digénisme (voir plus loin).
La difficulté de leur étude provient en partie des variabilités des phénotypes et des génotypes. Une même maladie peut apparaître si différents gènes sont mutés, des mutations différentes affectant le même gène peuvent donner des phénotypes identiques ou différents. Des facteurs environnementaux ou l'expression d'autre gènes semblent donc importants pour l'apparition de la maladie.
Dénominations
Le grand groupe des rétinopathies pigmentaires, maladies dégénératives de la rétine, correspond le plus souvent à une cohorte de patients qui présente une dégénérescence première des bâtonnets avec dégénérescence secondaire des cônes. On nomme classiquement cette dégénérescence retinitis pigmentosa (RP) ou rod-cones dystrophy (RCD).
Parfois seuls les cônes sont affectés, ce sont les les cones dystrophies (COD).
Les cônes peuvent être affectés en premier, ou secondairement à la dégénérescence des bâtonnets. Ce sont les cones-rod dystrophies (CRD).
La perte progressive des photorécepteurs amène généralement à la cécité en quelques dizaines d'années (excepté pour les COD) avec une grande variabilité de sévérité. La plus grave est l'amaurose congénitale de Leber (LCA).
Traitements
Actuellement il n'y a pas de traitement, mais on pense que l'on pourra dans l'avenir envisager des options thérapeutiques comme une thérapie génique ou une greffe de rétine.
On conseille le port de verres teintés pour lutter contre le soleil, Il semble en effet que certaines de ces pathologies soient aggravées par la lumière solaire. Il faut aussi envisager le traitement des lésions associées (cataracte, oedème maculaire).
La voie de la neuro-protection est ouverte car on pense que la mort des cônes qui suit la disparition des bâtonnets est secondaire à la disparition d'un facteur de survie. Plusieurs molécules sont efficaces chez l'animal, mais il faut encore de longues études pour évaluer leur efficacité chez l'homme et leur absence de nocivité.
Les thérapies géniques ou cellulaires sont en cours d'expérimentation chez l'animal et chez l'homme, et malgré quelques conséquences malheureuses (hémopathies des bébés-bulle), la voie est ouverte.
Les greffes de rétine ne consistent pas à changer la rétine pour recouvrer une vision normale. Les ophtalmologistes grefferont des fragments de rétine pour que l'ensemble des cellules visuelles ne meurent pas. Cela permettra sans doute de préserver les cônes indispensables pour la vision car ils ont tendance à disparaître à la suite des bâtonnets.
Quand un jeune couple a un enfant atteint de rétinopathie pigmentaire, sa première question sera de savoir si les enfants à venir présenteront aussi cette pathologie, ou bien s'il s'agit d'une maladie ponctuelle.
Les ophtalmologistes travaillent en collaboration avec leurs collègues des services de génétique ophtalmologique qui existent en France (nous donnons les coordonnées plus loin, dans la page4).
Quand on diagnostique une rétinopathie pigmentaire chez un patient, il est important d'examiner la famille à la recherche de marqueurs cliniques de l'affection. Ces anomalies minimes signeront le caractère familial de l'affection, ce qui doit entraîner la création d'un arbre généalogique de la parentèle. Il permettra de mieux appréhender la transmission de l'affection et d'envisager un conseil génétique pour la descendance.
Les médecins prennent ainsi de grandes responsabilités car la génétique est une science très complexe. On a parfois la surprise de constater en étudiant les gènes de la famille de l'enfant atteint, que le père n'est en fait pas le vrai père... Il faudra des trésors de subtilités pour conseiller sans bouleverser la famille. Et ce phénomène n'est pas exceptionnel (entre 5 et 10% des cas).
2) Historique des rétinopathies pigmentaires
La découverte en 1989 du gène responsable de la RP d'une famille irlandaise, localisé sur le bras long du chromosome 3 marquait le point de départ de l'étude génétique et chromosomique des RP. Quelques temps après on découvrit une mutation dans le gène de la rhodopsine au sein d'une population de malades américains présentant une RP, ce qui permit de faire des progrès dans la compréhension de ces pathologies.
Depuis cette date, de nombreuses mutations ont été décrites, touchant principalement la rhodopsine, la périphérine (retinal degeneration slow RDS), les anti-métalloprotéinases, la geronyl-geronyl-transférase, l'ornithine amino acid-transferase, la phosphodiestérase...
3) Clinique et examens complémentaires
a) Retinitis pigmentosa:
Il s'agit d'une rétinopathie à bâtonnets prédominants (90 à 95% des cas):
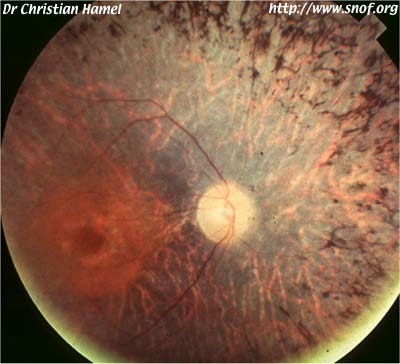
Rétinopathie pigmentaire
à bâtonnets prédominants RP
Le diagnostic est établi à l'occasion d'un bilan ophtalmologique pour une baisse de vision, une héméralopie ou bien un bilan familial devant la découverte d'un cas.
Le patient se plaint ainsi de ne plus rien voir dès que l'obscurité tombe, c'est l'héméralopie (du grec héméra le jour, et ops la vision). Cela est dû au dysfonctionnement des bâtonnets qui servent à la vision crépusculaire (night blindness).
Une photophobie surviendra plus tard.
Puis survient la perte progressive du champ visuel périphérique, dans un délai très variable par rapport au début de l'héméralopie.
La vision centrale peut rester très convenable pendant de longues années, mais on constate, à l'examen du champ visuel, un rétrécissement en canon de fusil (le patient ne voit que devant lui et ne voit rien autour). L'entourage s'en rend compte car il se cogne en marchant dans la rue, ne voyant pas les obstacles présents sur les trottoirs. Cet aspect évolué du champ visuel ne se retrouve pas au début de la maladie, et le champ visuel peut être tout à fait normal pendant des années.
Les signes cliniques sont fonction du type de transmission de la rétinopathie pigmentaire. Ainsi les formes autosomiques dominantes sont les moins graves, alors que les formes récessives ou liées à l'X sont graves.
A l'examen ophtalmologique on décrit des lésions bilatérales:
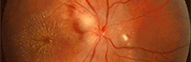
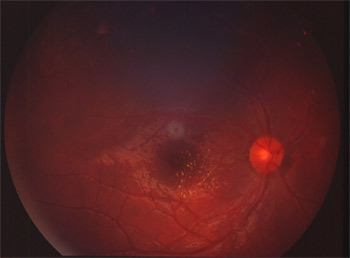
- une dépigmentation de l'épithélium pigmentaire, qui prend un aspect poivre et sel au début de la maladie. Puis le déplacement des cellules pigmentées va donner un aspect particulier de la rétine:
- une pigmentation périphérique de la rétine, avec un pigment plus ou moins concentré, formant des amas que l'on a comparé à des ostéoblastes.
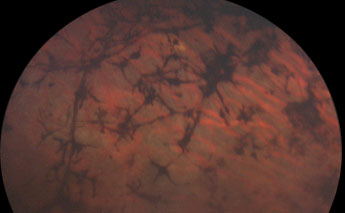
Pigmentation périphérique de la rétine (ostéoblastes)
Photo Pr Mathis CHU Rangueil Toulouse France
- des artères grêles sur tout le fond d'oeil
- une pâleur papillaire
- parfois une maculopathie avec oedème maculaire cystoïde ou atrophie maculaire.
- des signes moins constants peuvent être remarqués, que ce soit une cataracte postérieure, une myopie, un kératocône, un glaucome, des drusen papillaires, ou un vitré anormal.
Il y a une évolution des signes rétiniens, avec un aspect quasi normal au début, jusqu'à une pigmentation majeure quand la RP est évoluée. Il faudra donc s'attacher à bien rechercher les moindres signes chez le maximum de membres de la famille quand un cas a été diagnostiqué au sein de cette famille.
L'OCT (Ocular Coherence Tomography) commence à être utilisé et permet de mesurer de façon non invasive l'épaisseur de la rétine qui diminue lorsque l'on est face à une véritable RP. Elle est surtout utilisée pour le suivi d'une RP mais on s'en sert aussi pour un dépistage, nonobstant le prix élevé de ce matériel (90.000 euros).
Electrophysiologie:
L'électrorétinogramme (ERG) est anormal dès le début de la maladie. C'est un examen capital qui montre une diminution de l'onde b photopique et scotopique. Le délai d'apparition de l'onde b est aussi augmenté. Au début de la maladie on note seulement une altération de la composante scotopique (bâtonnets), puis une altération des systèmes photopiques et scotopiques, pour aboutir à un ERG "éteint".
L'ERG reste de très loin le moyen le plus sûr en complément de l'examen ophtalmologique avec étude du champ visuel et de la vision des couleurs. L'ERG multifocal devient de plus en plus utilisé et permet de mieux évaluer les fonctions maculaires.
"Seul l'ERG permet de distinguer entre un processus dégénératif stationnaire ou bien lentement évolutif , et un processus dégénératif évolutif. Seul l'ERG permet d'affirmer de façon souvent présymptômatique s'il y a une atteinte secondaire des cônes dans une rod-cone dystrophy (qui correspond à une RP) ou s'il n'y a qu'une atteinte des bâtonnets. L'ERG est donc indispensable" précise le Dr Abitbol du CERTO (Centre de Recherche Thérapeutique en Ophtalmologie).
Dans les formes liées à l'X, les réponses électrophysiologiques des cônes et des bâtonnets sont altérées. Dans certaines familles ayant une RP dominante avec une faible pénétrance, le tracé ERG des bâtonnets peut être réduit en amplitude et en latence, alors que la composante des cônes est normale.
L'électro-oculogramme (EOG) et les Potentiels Evoqués Visuels (PEV) sont souvent très utiles pour mieux cerner la pathologie.
L'adaptométrie est également un examen fondamental.
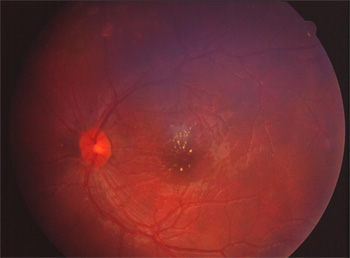
b) Cones Rod Dystrophies
Ce sont les rétinopathies à cônes prédominants (5 à 10% des cas):
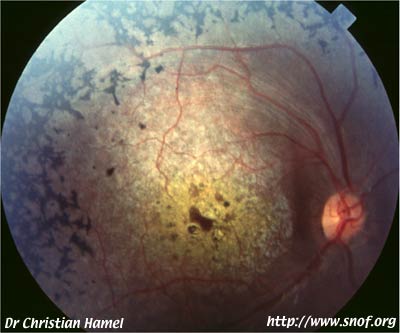
Rétinopathie pigmentaire
à cônes prédominants
Les cônes vont être atteints en premier, donc il va y avor une "inversion" de la symptomatologie, avec une altération primaire de la fonction visuelle, donc une baisse d'acuité visuelle, une photophobie et une dyschromatopsie précoces.
Le champ visuel met en évidence un scotome central, et l'ERG montre une diminution dramatique des ondes a et b avec altération prédominante de la fonction photophique.
Des dépôts pigmentaires apparaissent au niveau maculaire, associés à une atrophie vasculaire et une pâleur papillaire.
Ce n'est que plus tard que surviendront l'héméralopie, l'altération périphérique du champ visuel et du tracé scotopique de l'ERG.
5) Evolution
Une grande majorité de patient garde longtemps une vision utilisable pour leurs déplacements, malgré un champ visuel très étroit de 2 à 3 degrés. La conduite automobile est déconseillée car très dangereuse. Après 50 ans les malades gardent une vision très faible et peuvent même devenir aveugles.
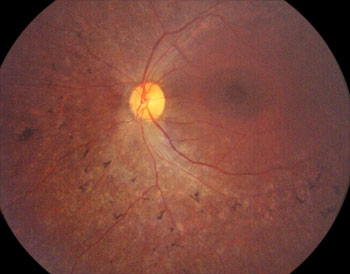
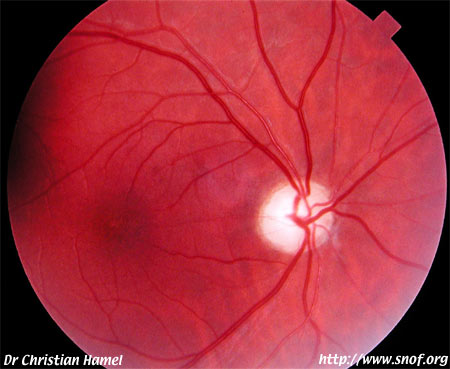
Rétinopathie pigmentaire
Photo Pr Mathis CHU Rangueil Toulouse France6) Notions de génétique
Cette spécialité médicale s'enrichit tous les jours de façon exponentielle. On décrit de très nombreux gènes en cause dans toutes les maladies génétiques, ce qui nous permet de mieux comprendre la fonction visuelle.
Nous encourageons donc le lecteur à se reporter aux nombreux ouvrages de génétique qui existent.
Le support de l'hérédité correspond aux chromosomes qui sont dans chaque noyau des cellules de l'organisme. Ils sont porteurs de gènes (50.000 à 100.00) qui sont des unités d'information responsables de la formation de l'ARN qui va entraîner la création de protéines (ensemble d'acides aminés). Ces gènes vont entraîner des phénotypes (du grec phainomai j'apparais), c'est à dire des caractères hérités des individus. On décrit des phénotypes pour la couleur des yeux (verts, bleus...), pour la myopie... L'ensemble des gènes responsables d'un phénotype donné est le génotype. Si le même caractère est présent sur le chromosome issu du père et de la mère, on dit que le sujet est homozygote pour ce caractère. Si le caractère est différent sur les deux chromosomes, il est dit hétérozygote.
Les différents variants d'un même gène sont appelés des allèles.
Certains phénotypes sont dits dominants quand le caractère génétique s'impose: le caractère "yeux marron" est dominant car les sujets hétérozygotes yeux marron/yeux bleus, ont des yeux marron. Le caractère yeux bleus est dit récessif. Il faut des sujets homozygotes yeux bleus/yeux bleus pour avoir des yeux bleus.
Les gènes sont répartis sur les 23 paires de chromosomes et sont séparés par des régions d'ADN non codant. Leur fonctionnement est régulé par des segments d'ADN.
L'information codante d'un gène correspond à des segments d'ADN appelé exons, séparés par des séquences non codantes appelées introns.
Les gènes sont organisés en groupes (clusters), ce sont les familles multigéniques simples qui regroupent des gènes identiques, et les familles multigéniques complexes qui contiennent des gènes semblables mais pas identiques.
Une altération d'un gène correspond à une mutation:
Les mutations ponctuelles :
- les missense mutations (faux-sens) correspondent à la modification d'un seule base, ce qui entraînera un changement de l'acide aminé produit (par exemple une lysine à la place d'une arginine), parfois sans incidence clinique, parfois donnant une pathologie.
- les nonsense mutations (non-sens) correspondent au changement du codon d'un acide aminé en un codon stop qui va stopper la fabrication de la protéine. Elle va donc se retrouver plus courte, ayant perdu une partie de sa région carboxy terminale. Cela donne un effet important sur la protéine finale et produira souvent un phénotype mutant (un patient malade).
- les frameshift mutations (déphasage) correspondent à l'insertion de bases surnuméraires ou à la délétion de bases existantes. Un phénotype mutant apparaîtra très souvent.
- les silent mutations (silencieuses) n'ont pas d'effet sur la protéine et n'entraînent rien pour le patient. Quand une base purique est remplacée par une base pyrimidique il s'agit d'une transversion, et quand il s'agit du remplacement d'une base pyrimidique par une autre ou d'une base purique par une autre, il s'agit d'une transition.
Les mutations de grande ampleur :
- les délétions correspondent à la perte d'une portion d'ADN, plus ou moins importante
- les insertions correspondent à l'insertion de bases surnuméraires venant habituellement d'une autre partie du chromosome
- les réarrangements correspondent à des échanges mutuels de segments d'ADN à l'intérieur ou à l'extérieur d'un gène
Ces mutations donnent toujours un phénotype pathologique.
On décrit différentes transmissions des caractères héréditaires:
Les hommes possèdent les chromosomes sexuels XY et les femmes possèdent XX. Les allèles récessifs ne s'expriment pas chez les femmes hétérozygotes XmX; elles vont transmettre le gène anormal à la moitié de leurs filles XmX et auront une moitié de garçons atteints de la maladie XmY. Ces hommes n'ayant qu'un X (hémizygotes) sont nécessairement homozygotes pour X et vont exprimer leurs allèles récessifs. Ceci est visible pour les dyschromatopsies. Les femmes transmettent l'anomalie de leur chromosome X, elles sont conductrices, et les hommes présentent la dyschromatopsie. On décrit aussi le rétinoschisis juvénile. Parfois les femmes conductrices XmX ont quelques signes cliniques à cause de la lyonisation. Ce phénomène normal correspond à l'inactivation d'un chromosome X, laissant l'autre fonctionnel. Si par malchance on a une inactivation du X sain, le X malade pourra s'exprimer et entraînera des signes pathologiques plus ou moins importants.
Il faudra s'attacher à bien examiner la mère et les soeurs de ces garçons atteints. Par exemple quand on examine une femme conductrice de la maladie de Fabry (déficit en alpha galactosidase) on constate une cornée verticillée (cornea verticillata) comparable à ce qu'on voit lors d'une thésaurismose d'amiodarone. Cela signe le caractère familial et l'hétérozygotie de la mère.
Les rétinopathies pigmentaires de cette catégorie sont celles qui ont le plus mauvais pronostic et représentent 8% des rétinopathies pigmentaires.
Il existe des transmissions du père au fils de certains caractères présents sur le chromosome Y; c'est la transmission holandrique.
- Transmission autosomique dominante
les caractères génétiques sont transmis par les chromosomes non sexuels (autosomiques) sur un mode dominant.
Bien qu'hétérozygote, le sujet exprimera la maladie. Ces rétinopathies sont d'assez bon pronostic et sont les plus fréquentes (43% des cas)
- Transmission autosomique récessive
les caractères génétiques sont transmis par les chromosomes non sexuels (autosomiques) sur un mode récessif. Il faut que le sujet soit homozygote pour que la pathologie apparaisse. Ces rétinopathies correspondent à 20% des cas environ.
- Transmission mitochondriale
Les mitochondries sont des organelles contenues dans le cytosplasme cellulaire. On admet qu'elles correspondent à des organismes procaryotes qui ont été "captés" par les cellules eucaryotes, il y a très longtemps. Le matériel génétique de ces organites provient toujours de la mère car les spermatozoides n'en contiennent pas. L'article du 22 août 2002 apporte peut-être un nouvel éclairage sur cette transmission:Paternal Inheritance of Mitochondrial DNA Schwartz M., Vissing J. N Engl J Med 2002; 347:576-580, Aug 22, 2002.
Certaines pathologies sont dues à des mutations de cet ADN mitochondrial, comme l'atrophie congénitale de Leber.
Le digénisme diallélique signifie que pour que la maladie se développe, il est nécessaire d'avoir une mutation dans deux gènes différents. C'est une situation qui est probablement relativement fréquente mais encore très mal connue. Dans le cas du digénisme RDS + ROM1, il s'agit d'une découverte faite par le groupe de Thaddeus Dryja à Harvard. Ce groupe avait déjà découvert chez plusieurs familles des mutations à l'état hétérozygote dans le gène RDS et, pour certaines familles, un certain nombre d'individus mutés ne développaient pas la maladie. Par la suite, en recherchant systématiquement des mutations dans le gène ROM1 (découvert après RDS) chez tous les patients dont ils avaient l'ADN (que ceux-ci aient des mutations dans un autre gène ou non), ils ont découvert que certains ayant des mutations dans RDS avaient aussi des mutations dans ROM1. Ils se sont aperçus que les patients n'ayant des mutations que dans l'un des deux gènes n'étaient pas malades. Par contre ceux qui étaient hétérozygotes pour une mutation de chacun des deux gènes l'étaient. Ainsi, l'explication de l'absence de maladie initiale chez certains patients mutés dans RDS était que ces patients n'avaient pas de mutations dans ROM1. Attention cependant, seules quelques mutations RDS répondent à ce schéma. La plupart des mutations RDS suffisent à elles seules pour entraîner la maladie.
On décrit un digénisme triallélique BBS2/BBS6 et BBS4/BBS2 dans le syndrome de Bardet-Biedl. Il faut deux mutation sur un allèle plus une sur un autre allèle pour que la maladie apparaisse. Le texte en fichier pdf de 1,5M "Beyond Mendel : an evolving view of human genetic disease transmission"  de Jose L. Badano and Nicholas Katsanis est très intéressant à ce sujet.
de Jose L. Badano and Nicholas Katsanis est très intéressant à ce sujet.
7) La rhodopsine

Bâtonnet
Les cellules visuelles de la rétine sont les cônes et les bâtonnets.
Les cônes sont 3 millions dans chaque rétine, concentrés au niveau de la macula, et servent à la vision fine et à la vision des couleurs.
Les bâtonnets en revanche sont beaucoup plus nombreux, environ 100 millions par oeil, et servent à la vision crépusculaire, dès que la lumière diminue. Ils présentent une extraordinaire sensibilité puisqu'un seul photon suffit à déclencher la cascade biochimique de la vision (phototransduction).
Le bâtonnet est une cellule rectiligne et est composé de deux parties, le segment externe d'environ 40 microns et le segment interne plus petit qui contient le noyau et les organelles.
Le segment externe contient environ 2000 disques empilés et très proches les uns sur les autres, formés d'une membrane dérivée de la membrane plasmique. C'est dans la membrane des disques qu'on trouve la molécule sensible à la lumière, la rhodopsine. Il faut noter que la membrane cellulaire (plasmique) est distincte de la membrane entourant les disques.
Le segment interne contient les organites nécessaires à la formation des molécules rentrant en jeu dans le phénomène de la vision. Il sert au remplacement des molécules qui sont situées dans le segment externe, et comprend aussi la synapse qui est la connexion avec les autres cellules de la rétine.
Quand un photon est capté par les molécules des disques, de nombreux phénomènes biochimiques vont se produire, pour transformer l'énergie lumineuse en une énergie électrique qui sera transmise le long de la membrane plasmique jusqu'à la terminaison synaptique, vers les autres cellules de la rétine.
Les étapes de la phototransduction (transformation de la lumière) sont assez bien connues actuellement pour les bâtonnets, contrairement à ceux des cônes.
Phototransduction dans le bâtonnet
La rhodopsine des disques est un photopigment qui est composée de deux parties, une partie protéique, l'opsine et un groupement photophore, l'isomère 11-cis du rétinal (c'est l'aldéhyde de la vitamine A).
L'opsine est une molécule de 348 acides aminés et est formée de 7 hélices alpha reliées par des segments polypeptidiques. L'isomère 11-cis se situe à l'intérieur de la molécule et est relié à une des hélices alpha. Il est formé de 6 atomes de carbone et une chaîne latérale dite conjuguée, car il y a alternance de liaison simples et doubles.
L'isomère cis est caractérisé par une chaîne latérale coudée entre les atomes de carbone 11 et 12.
Lorsque la lumière éclaire la rétine, on assiste à une transformation du cis-rétinal en tout-trans-rétinal, sans coude au niveau de sa chaîne conjuguée. La liaison avec l'opsine devient instable et va s'hydrolyser. On assiste alors au passage de la molécule en métarhodopsine I puis métarodhopsine II, avec émission d'un message neuro-chimique à ce moment-là.
Il suffit d'un photon pour qu'une molécule soit isomérisée et transmette l'information au cerveau.
A l'obscurité, le tout-trans-rétinal va être modifié par une enzyme isomérase qui va le transformer en 11-cis rétinal. Il va être capté par l'opsine pour reformer une nouvelle molécule de rhodopsine.
L'activation de la rhodopsine par la lumière entraîne, par l'intermédiaire de protéines G spécifiques, la modification du taux de GMP cyclique. Ce GMP cyclique (ou 3'5'guanosine monophosphate cyclique) commande un canal sodium.
Dans le noir les niveaux de GMP cyclique sont hauts et le canal est ouvert, la membrane est dépolarisée et la synapse active, largant des neurotransmetteurs.
A la lumière, l'activation de la rhodopsine entraîne l'activation d'une enzyme, la transducine, formée de trois sous-unités, alpha, bêta et gamma. La sous-unité alpha va voir son GDP remplacé par un GTP, et va se lier à une enzyme phosphodiestérase. Elle va entraîner avec elle la sous-unité inhibitrice gamma de la phosphodiestérase. Ce départ active la phosphodiestérase qui va ouvrir, hydrolyser 4200 molécules de GMP cyclique par seconde, la GMP cyclique se transformant en 5'GMP. Donc cette GMPc devient rare, ce qui va fermer le canal sodium, ce qui stoppe l'émission du neurotransmetteur. Cette information (hyperpolarisation) est relayée par un jeu complexe de cellules bipolaires et de cellules horizontales.
Quand la GTP de la sous-unité alpha de la transducine s'hydrolyse en GTP et en phosphate libre, cette sous-unité libère l'unité inhibitrice gamma de la phosphodiestérase qui se recombine à ses deux sous-unités alpha et bêta, et remet l'enzyme au repos. Une enzyme, l'arrestine, va bloquer la fixation de la transducine sur la rhodopsine et la remettre dans les conditions d'obscurité.
Il faut noter qu'on a assisté à une amplification du phénomène car une molécule de rhodopsine activée par un photon va donner naissance à des centaines de complexes actifs de transducine. L'étape suivante, l'activation de la phosphodiestérase, se fait sans amplification, molécule pour molécule. Mais l'étape suivante amplifie encore le processus puisqu'une molécule de phosphodiestérase hydrolyse plusieurs milliers de molécules de GMP cyclique.
Ces réactions sont modulées par de nombreuses autres protéines comme la rhodopsine kinase, ou la recoverine. Deux catégories de protéines sont importantes pour la stabilité du bâtonnet, la ROM1 (Rod Outer segment Membrane protein 1) et la périphérine/RDS. Une mutation dans un des gènes responsable de leur fonctionnement entraînera d'importantes anomalies de fonctionnement du bâtonnet.
8) Biologie moléculaire
Dès les années 1920, C.Keeler à l'Université Harvard trouva que certaines souris avaient des déficits en bâtonnets rétiniens, animaux qu'il appela "rodless" ou "r". Plus tard dans les années 1950, on découvrit les souris "rd" (retinal degeneration) maintenant appelée rd1. Une étude par PCR montra que les deux lignées avaient le même gène déficient, Y347ter dans le gène PDEß. Plus tard dans les années 30, on a décrit un type de setter irlandais porteur de gène rcd1, au niveau W807ter (correspondant à la souris). Plus tard encore, dans les années 70 on trouva la souris rds (rd2).
C'est le gène codant la glycoprotéine périphérine/rds qui est responsable de dégénérescence rétinienne retrouvée chez la souris rds (rds/peripherin).
La périphérine est donc cette protéine qui forme le segment externe des photorécepteurs. Les souris rds ne peuvent pas synthétiser cette protéine, ce qui va donner une dégénérescence rétinienne.
Les premiers patients qui ont été étudiés présentaient au niveau du bras long du chromosome 3 une mutation proline-histidine sur le codon 23 de la rhodopsine. Ces malades avaient une RP automosomique dominante. Les mutations décrites au niveau de cette molécule sont responsables d'environ 30% des RP autosomiques dominantes.
Chez l'homme une mutation faux-sens du codon 150 a été associée à avec une rétinopathie pigmentaire autosomique récessive. De même pour le gène codant la subunité bêta de la phosphodiestérase (PDE).
Le Dr Hamel évoque les possibilités d'une future thérapie génique, lors du Congrès de la Société Européenne de Thérapie Génique (octobre 2000):
"Jusqu'alors des essais ont été faits avec succès dans deux modèles de rétinite pigmentaire autosomique dominante avec AAV[virus associé aux adénovirus], les souris rds et les rats transgéniques P23H, et dans un modèle de rétinite pigmentaire autosomique récessive avec HIV1, les souris rd. Chez le rat par exemple, le traitement permet de préserver plus de 50% des bâtonnets après un an, ce qui représente la moitié de la longévité de l'animal, sachant que sans traitement les bâtonnets ont totalement disparu à cet âge-là. Des essais sont en cours maintenant pour voir si la même efficacité est obtenue avec des yeux d'une taille semblable à celle de l'homme.
Compte tenu des résultats obtenus jusqu'à maintenant, on peut penser que les premiers essais chez l'homme commenceront dans 2 à 3 ans. Il faudra alors encore quelques trois à cinq ans pour s'assurer de l'innocuité et de l'efficacité de ce traitement avant de le proposer aux malades dans un cadre non expérimental.
Ceci ne doit pas nous faire oublier que d'autres stratégies thérapeutiques sont à l'essai, notamment celles qui visent à protéger les cônes. Ces approches ont pour but d'apporter des médicaments directement dans l'œil, à proximité des photorécepteurs, ou consistent à effectuer des greffes de bâtonnets. Là aussi quelques petites années seront encore nécessaires pour dégager clairement les indications. Il nous faut donc être encore un peu patients, mais nous savons que ceci ne sera pas vain. "
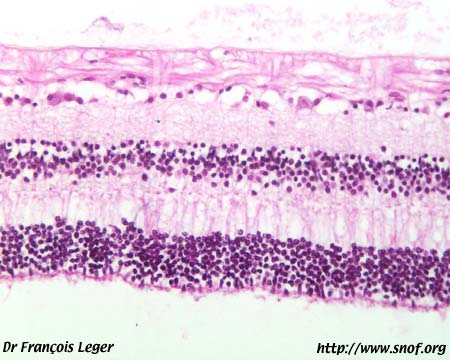
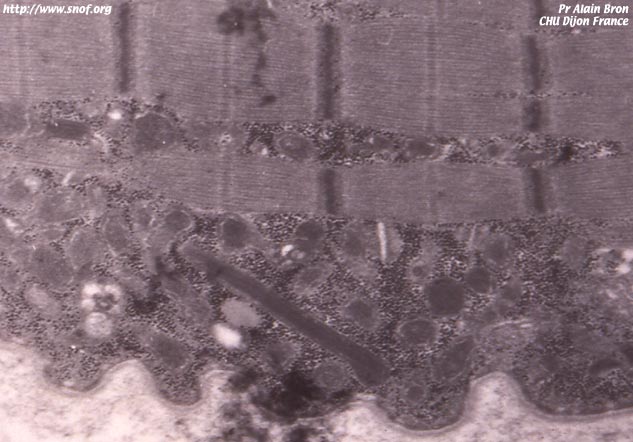
Coupe histologique de rétine adulte normale9) Classement des dystrophies héréditaires de la rétine
Dystrophies héréditaires de la rétine
- Rétinopathies pigmentaires non syndromique (65% à 85% des cas suivant les auteurs)
- Rétinopathies pigmentaires associées à des syndromes généraux:
- Syndrome de Bassen-Kornzweig (abêtalipoprotéinémie, avec ataxie progressive, stéatorrhée, et rétinopathie poivre et sel)
Il s'agit d'une abêtalipoprotéinémie familiale, autosomique récessive, due à une mutation du gène situé en 4q22-q24. Elle associe un syndrome coeliaque, une RP, une ataxie progressive, et l'existence de globules rouges ayant une forme spéciale, spiculés, appelés acanthocytes. L'absence de bêtalipoprotéine dans le sang correspond à l'impossibilité de synthétiser le peptide apoB des LDL et des VLDL.
L'hypocholestérolémie importante s'accompagne d'une malabsorption intestinale des vitamines liposolubles A, E et K donnant les signes généraux. Une démyélinisation centrale et périphérique résulte de cette anomalie et s'ajoute à une stéatose hépatique parfois transformée en cirrhose.
Bassen, F. A.; Kornzweig, A. L. : Malformation of the erythrocytes in a case of atypical retinitis pigmentosa. Blood 5: 381-387, 1950.
- Dystrophie cristalline de Bietti
- Maladies périxosomiales
- Syndrome de Zellweger (cérébro-hépato-rénal)
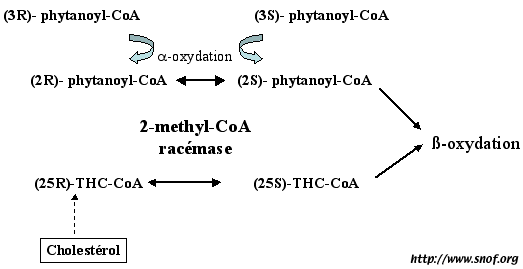
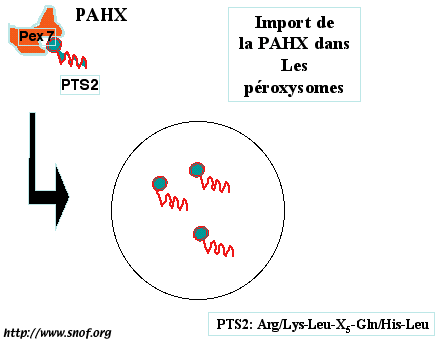
- Syndrome de Refsum
- Adrénoleucodystrophies
- Hyperoxalurie type 1
- Cystinose
- Lipofuscinose (retard mental, ataxie, macula rouge cerise)
- Ataxie cérébelleuse de type II (SCA7) autosomique dominante
- Dystrophie myotonique
- Syndrome de Hallervorden-Spatz (dysarthrie progressive et démence, maculopathie en oeil de boeuf)
- Syndrome de Joubert (hypoplasie du vermis, kystes rénaux)
- Syndrome de Usher (12% des cas)
- Syndrome de Kearns-Sayre
- Mucopolysaccharidoses
- Syndrome de Bardet-Biedl (CRD, retard mental, polydactylie, obésité et hypogonadisme, avec parfois malformations rénales. Au moins 7 gènes sont responsables avec des cas de digénisme triallélique)
- Syndrome de Alström (idem sans polydactylie)
- Syndrome de Laurence-Moon à ne pas confondre avec le Bardet-Biedl, (rétinite pimentaire, paraplégie spastique et retard de taille)
- Ataxie de Friedreich
- Myopathie de Duchenne
- Syndrome de Senior-Loken (sévère RP, parfois diagnostiqué LCA, anomalies rénales nécessitant souvent une transplantation rénale).
- Syndrome d'Alport (surdité, néphropathie progressive, tâches jaunâtres autour de la macula)
- Syndrome de Cohen (dysmorphie faciale, petite taille, retard mental, longues mains, neutropénie)
- Syndrome de Jeune (hypoplasie thoracique, brachidactylie, néphropathie)
- Syndrome de Cokraine (nanisme, progeria, retard mental, et rétinopathie avec fines ponctuations)
- Héméralopies
- Héméralopie de type Nougaret
- Héméralopie de type Schubert-Bornstein
- Maladie d'Oguchi
- Fundus albipunctatus cum hemeralopia de Lauber
- Carence en Vitamine A
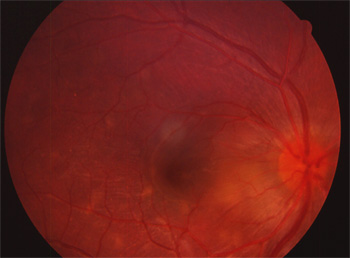
- Amaurose congénitale de Leber (5% des cas)
- Drusens familiales dominante
- Maculopathies
- Dystrophie des cônes (3% des RP)
- Pattern dystrophies
- Dystrophie maculaire de la Caroline du Nord
- Dystrophie de Sorsby
- Maladie de Best (1% des cas)
- Maladie de Stargardt (6% des cas)
- Dystrophies choroïdiennes
- Choroïdérémie (2% des cas des rétinopathies pigmentaires, anomalie lié au chromosome X)
- Atrophie gyrée (très rare, autosomal récessive)
- Atrophie choroïdienne diffuse
- Dystrophie aréolaire centrale
- Neuropathie optique héréditaire
- Vitréorétinopathies
- Rétinoschisis juvénile lié à l'X (1% des cas)
- Syndrome de Goldmann-Favre (récessif autosomique, associant une héméralopie à un rétinoschisis)
- Vitréorétinopathies héréditaires
- Vitréorétinopathie familiale exsudative
- Maladie de Wagner
- Syndrome de Stickler
- Maladies inflammatoires de l'oeil
- Myopie
- Albinisme oculaire et cutané
- Albinisme oculaire
- NARP (Neuropathy, Ataxia, and Retinitis Pigmentosa)
10) Transmission
Les cas sporadiques représentent 45% des cas.
Les 55% de cas familiaux se répartissent ainsi:
- Dominants autosomiques 20 %
- Récessifs autosomiques 20%
- Récessifs lié à l'X 14%
- Fratrie de garçons 1%.
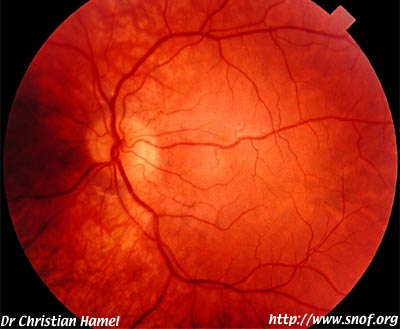
Albinisme oculaire pur
11) Mutations
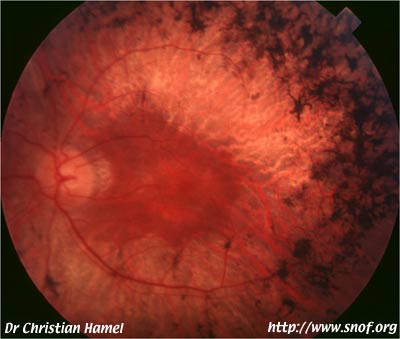
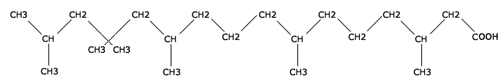
Mutation RDS P216S 11.1 Généralités
On trouve régulièrement des gènes responsables de rétinopathies pigmentaires ou de dégénérescences rétiniennes variées.
Le site de l'Ocular Molecular Genetics Institute (OMGI) décrit 108 gènes responsables à ce jour de dystrophies héréditaires de la rétine. L'ensemble des gènes est classé en fonction de leurs caractéristiques. RetNet comptabilise 139 gènes.
Nous en resterons aux mutations donnant des rétinopathies pigmentaires, ce qui est déjà considérable.
La génétique des rétinopathie pigmentaire est complexe, à cause de la variété des transmissions familiales, de l'hétérogénéité des phénotypes. La plupart de ces gènes sont exprimés dans les photorécepteurs, 4 encodent des éléments de la cascade de la phototransduction (rhodopsine, sous-unité a et b de la phosphodiestérase cGMP).
"L’hétérogénéité génétique de ces maladies est considérable, avec 39 gènes (31 clonés, 8 mappés) actuellement répertoriés répondant à 50 % des cas de RP non syndromiques et 10 gènes (6 clonés, 4 mappés) pour les CRD (cônes prédominants) non syndromiques, ce qui freine la mise en place d’un diagnostic moléculaire." Dr Hamel mai 2003.
Les mutations atteignent différentes étapes de la fonction visuelle:
les protéines de la transduction visuelle des bâtonnets:
- rhodopsine,
- subunités alpha et bêta de la phosphodiestérase,
- subunités alpha et bêta du cGMP des canaux,
- l'arrestine,
les protéines du cytosquelette des bâtonnets:
- périphérine/RDS,
- ROM1,
- fascine,
- prominine like 1
les protéines probablement engagées dans les bâtonnets:
les protéines engagées dans la différentiation des photorécepteurs:
- NRL,
- facteur de transcription PNR
les protéines engagées dans la duplication de l'ARNm:
les protéines engagées dans le métabolisme des nucléotides:
les autres métabolismes:
- Tubby,
- tubby like protéine 1,
- CRB1,
- MITS2.
Par ailleurs, les RP peuvent être dues à des mutations des protéines du métabolisme du rétinol (comme ABCA4 qui est aussi en cause dans la maladie de Stargardt). Beaucoup d'autres molécules sont exprimées dans l'épithélium pigmentaire rétinien (RPE65, l'isomérase RGR lumière dépendante, le 11-cis retinoid transporter CRALBP ou le lecithin retinol acyl transferase LRAT.
Pour les CRD, les protéines sont exprimées dans les bâtonnets et dans les cônes, comme ABCA4, CRX (rôle dans la différenciation des photorécepteurs), le guanylate cyclase qui peut aussi donner une LCA, RIM1, HRG4 ou dans le épithélium pigmentaire rétinien (MERTK).
Nous ne présenterons que quelques mutations.
11.2 Classification génétique des rétinopathies pigmentaires
Les formes autosomiques dominantes (ADRP pour autosomal dominant RP) sont dues à des mutations des gènes:
- RHO (rhodopsin) à l'emplacement 3q21-24,
- RDS (RDS/peripherin) en 6p21.1-cen,
- RP1 en 8p11-21,
- RGR en 10q23,
- ROM1 (rod outer segment membrane protein 1) en 11q13,
- NRL (neural retina leucine zipper transcription factor) en 14q11.1-11.2,
- CRX en 19q13.3,
- PRKCG en 19q13.4
Les formes autosomiques récessives (ARRP pour autosomal recessive RP) correspondent à des mutations:
- RPE65 (retinal pigment epithelium 65) en 1p31: la plupart donnent des LCA (Leber congenital amaurosis),
- ABCA4 en 1p21-13,
- CRB1 en 1q31-32.1,
- USH2A en 1q41,
- MERTK en 2q14.1,
- SAG en 2q37.1,
- RHO en 3q21-24,
- PDE6B (beta subunit of rod cGMP-phosphodiesterase) en 4p16.3,
- CNGA1 (alpha subunit of the rod cGMP-gated cation channel) en 4p14-q13,
- PDE6A (alpha subunit of rod cGMP-phosphodiesterase) en 5q31.2-34,
- TULP1 en 6p21.3,
- RGR en 10q,
- NR2E3 en 15q23, et
- RLBP1 en 15q26
Les formes liées à l'X (XLRP pour X-linked RP) correspondent à des mutations:
Les formes consécutives à un digénisme:
- Mutations hétérozygotes de ROM1 en 11q13, associées à des mutations hétérozygotes de RDS en 6p21.1-cen
Les formes associées
- MYO7A avec syndrome de Ush1
- Bardet-Biedl
Le gène RPGRIP donne des LCA qui sont considérées comme une pathologie distante des RP.
11.3 Présentation des mutations principales
11.3.1 Rhodopsine (Rho) (RP4)
Le premier gène muté de la rhodopsine donnant une RP à transmission autosomique dominante (ADRP) fut découvert en 1989 dans une grande famille irlandaise, localisé sur le bras long du chromosome 2. Depuis cette date, plus de 100 mutations ont été décrites dans ce groupe ADRP.
En 1992 Dryja et al décrivirent la première mutation de la rhodopsine donnant une RP à transmission autosomique récessive (ARRP).
Humphries et al, en 1997, créèrent des souris knockout (souris à qui on a enlevé un gène) porteuses de mutations de la rhodopsine. Les bâtonnets des souris rho-/- ne contenaient pas de segments externes, alors que les animaux hétérozygotes rho-/+ avaient des segments petits et désorganisés. Les hétérozygotes avaient des ERG normaux. Ce type de souris knockout devint un animal intéressant pour étudier les RP dues à des mutations du gène de la rhodopsine.
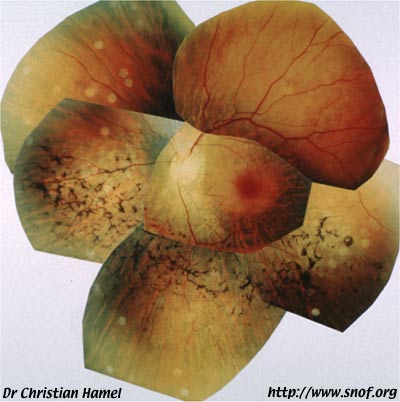
Mutation de la rhodopsine RHO G06R
Des études ont mis en évidence des mutations particulières du gène RHO, comme par exemple la mutation E341X chez des chinois, avec un changement du codon 341 (changement de GAG à TAG). Cette mutation est due à un codon stop aboutissant à une protéine de rhodopsine tronquée. Les maladies présentent une rétinopathie pigmentaire avec atrophie optique, héméralopie, et quelques ostéoblastes périphériques.
Une étude du gène RHO dans le sud de la France pour mesurer la prévalence des altérations de la rhodopsine, montra que ces mutations ne sont responsables que de 10.3% des ADRP dans cette région.
Bareil C, Hamel C, Pallares-Ruiz N, Arnaud B, Demaille J, Claustres M. Molecular analysis of the rhodopsin gene in southern France : identification of the first duplication responsible for retinitis pigmentosa, c.998^999ins4. Ophtalmic Genet 1999, 20:173-82.
11.3.2 Périphérine / RDS (RP7)
Cette molécule est fondamentale pour que la structure des segments externes des cellules visuelles soit normale.
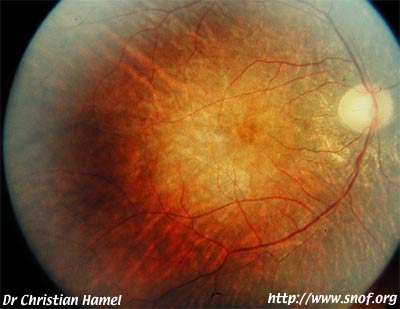
Différents phénotypes sont associés à cette anomalie, fundus flavimaculatus, dystrophie maculaire en ailes de papillon, dystrophie maculaire vitelliforme, pattern dystrophy.
Des souris présentant de telles mutations existent, ce sont les mutants rds (retinal degeration slow), présentant un déficit de développement de leurs segments externes, avec dégénerescence des photorécepteurs, que les souris soient homozygotes ou hétérozygotes, contrairement aux mutations de la rhodopsine comme on l'a vu.
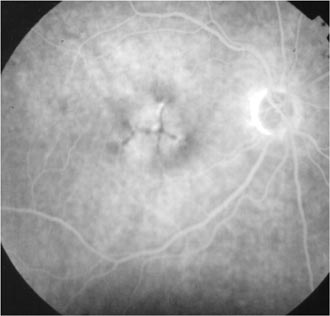
Dystrophie vitelliforme de l'adulte RDS G305D
ROM1 (rod outer segment membrane protein 1)
Ce gène a été localisé en 11q13, au niveau de la région responsable de la maladie de Best et du syndrome de Usher type 1.
RPE65 (retinal pigment epithelium 65)
Ce sont des protéines rentrant en jeu dans le métabolisme du rétinol. Le gène est surtout en jeu dans l'amaurose congénitale de Leber. On n'a décrit que de rares cas de rétinite pigmentaire. La protéine RPE65 fait partie du complexe moléculaire d'isomérisation. Les mutations CRALBP (cellular retinaldehyde binding protein) et LRAT donnent un phénotype similaire.
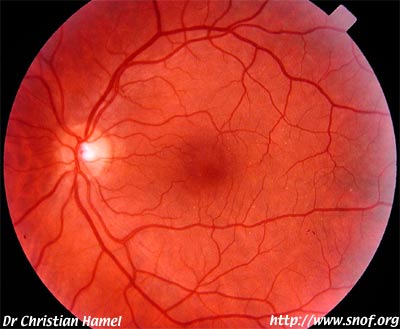
Mutation RPE65 hétérozygote

Mutation RPE65 hétérozygote
Gène RP1
Ce gène de 4 exons est spécifique de la rétine et est situé sur le chromosome 8.
Il code une protéine ORP1 oxygene-regulated protein (surexprimée en cas d’hypoxie). Il existe une homologie avec le gène DCX (migration des neurones au cours du développement). L'atteinte rétinienne peut être localisée en nasal inférieur. On note des variations de sévérité intrafamiliale importantes.
Fascine
Cette protéine est spécifique de la rétine. Elle est impliquée dans l’assemblage des filaments d’actine dans le cil connecteur. On a seulement décrit une mutation dans 4 familles japonaises.
Neural Retine Zipper
C'est un facteur de transcription spécifique de la rétine. Il active la transcription du promoteur de la rhodopsine en synergie avec le gène CRX. Seulement 1 mutation dans 3 familles britanniques
RP2
Ce gène de 5 exons, n'est pas spécifique de la rétine. Il existe une homologie avec le cofacteur C qui est impliqué dans le repliement de la tubuline b, nécessaire à son hétérodimérisation avec la tubuline a. Ce sont des formes moins sévères que RP3.
RPGR
Le facteur Retinitis pigmentosa GTPase regulator, régule et stabilise le GTP.
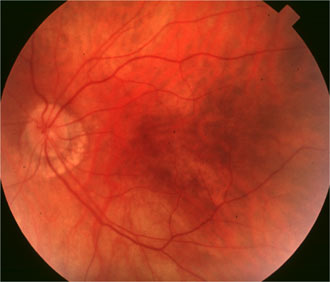
Mutation RPGR Mère de 65 ans |
Mutation RPGR Fils de 35 ans |
On a décrit une mutation liée à l'X dans une famille chinoise, par délétion 28-pp dans l'exon 7 à cause d'un codon stop qui a éliminé la terminaison C de la protéine RPGR. Les hommes de la famille présentaient une rétinopathie pigmentaire alors que les femmes étaient myopes, avec des anomalies de l'ERG. Kanxing Zhao, Lejin Wang, Li Wang , Liming Wang, Qingsheng Zhang et Qing Wang Novel deletion of the RPGR gene in a Chinese family with X-linked retinitis pigmentosa Opthalmic Genetics 2001, Vol.22, No.3, pp. 187-194
Protéine RIM et gène ABCR (ATP Binding Cassette Receptor)
Le gène ABCR est muté chez 1/50 à 1/80 personnes. On le retrouve dans la maladie de Stargardt mais aussi dans des rétinites pigmentaires et cone rod dystrophy.
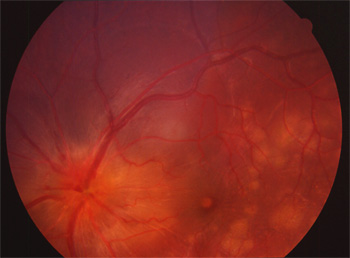
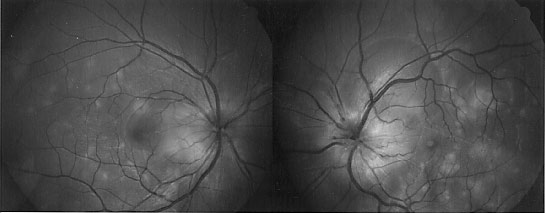
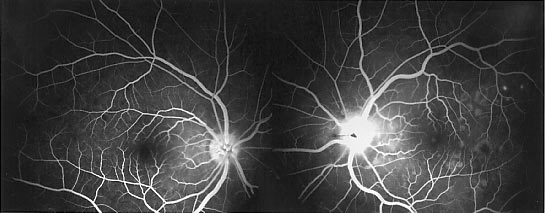
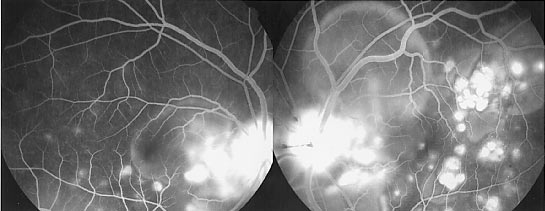
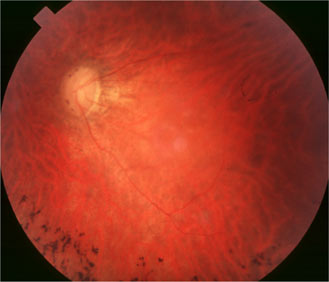
Protéine RDH5
Elle fait partie du complexe moléculaire d’isomérisation.
Les phénotypes correspondent à un Fundus albipunctatus et une dystrophie des cônes.
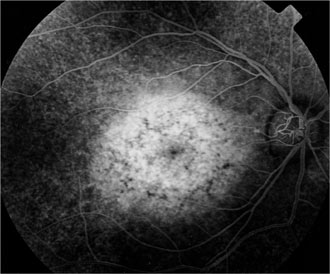
Mutation RDH5
TULP1
Les rétinites pigmentaires sont sévères à cause d'une anomalie de l’opsine des cônes et bâtonnets.
MERTK
Ces rétinites pigmentaires sévères peuvent êtres associées à une polydactylie. Elle sont dues à un défaut de phagocytose de la part de l’épithélium pigmentaire. On possède un modèle du rat RCS.
NR2E3
Ces rétinites pigmentaires ou enhanced-S cone syndrome correspondent à une augmentation des cônes bleus.
CRB1
Ce sont des rétinites pigmentaires sévères ou une amaurose congénitale de Leber.
RP13
Une famille allemande présentant une rétinite pigmentaire autosomique dominante (ADRP) correspondait à une mutation missense dans l'exon 42 du gène RP13, fut décrite:
J.J.C. van Lith-Verhoeven, S.D. van der Velde-Visser, M.M. Sohocki, A.F. Deutman, H.M.A. Brink, F.P.M. Cremers et C.B. Hoyng Ophthalmic Genetics 2002, Vol.23, No.1, pp. 001-012
12) Consultations de génétique ophtalmologique
Dr Pierre BITOUN
Consultation de génétique ophtalmologique
Hôpital Jean Verdier
Avenue du 14 juillet
93140 BONDY
FRANCE
Téléphone : 01 48 02 64 62
Fax : 01 48 02 68 23
Pr Jean-Louis DUFIER
Dr Olivier ROCHE
Consultation d'ophtalmologie adultes et enfants
Hôpital Necker - Enfants Malades
149 Rue de Sèvres
75743 PARIS CEDEX 15
FRANCE
Téléphone : 01 44 49 40 00
Fax : 01 47 34 72 09
Dr Christian HAMEL
Consultation de dystrophies héréditaires de la rétine
Service d'ophtalmologie
Hôpital de Gui de Chauliac
80, avenue Auguste Fliche
34295 MONTPELLIER cedex 05
Tél 04 67 33 72 53
Dr Josseline KAPLAN
Service de génétique médicale
Hôpital Necker - Enfants Malades
149 Rue de Sèvres
75743 PARIS CEDEX 15
FRANCE
Téléphone : 01 44 49 51 62
Fax : 01 44 49 51 50
Dr Pierre BITOUN
SIDVA 91
95 Rue Roger Salengro
91600 SAVIGNY-SUR-ORGE
FRANCE
Téléphone : 01 69 12 25 50
Fax : 01 69 12 25 51
Dr Hélène DOLLFUS
Clinique ophtalmologique
CHU Hôpital de Hautepierre
Avenue Molière
67098 STRASBOURG CEDEX
FRANCE
Téléphone : 03 88 11 65 67
Fax : 03 88 11 63 41
13) Conclusion
Actuellement nous ne réalisons que peu de diagnostics anténataux, car il faudrait savoir quelle mutation chercher, et sur quel gène. Il y a tout de même les familles présentant une RP liée au chromosome X qui peuvent en bénéficier. Si on arrive à démontrer cette transmission, on pourra étudier les deux principaux gènes en cause qui sont RPGR (RP3) qui représente 25 à 30% de ces familles, et RP2 qui en représente 10 à 15%. En cas d'identification d'une mutation on pourra proposer un diagnostic anténatal.
L'avenir devrait nous permettre de mieux comprendre le fonctionnement des gènes et leur retentissement sur la vision. Il s'agit là d'un travail important mais qui est à notre portée.
14) Avenir
Une nouvelle piste thérapeutique pour la rétinite pigmentaire (Communiqué Inserm - 28 juin 2004)
Une équipe de chercheurs français, dont les travaux seront publiés dans la revue Nature Genetics de juillet, a identifié une nouvelle protéine nécessaire à la survie de certains photorécepteurs, essentiels à l'acuité visuelle. Des bâtonnets ont été transplantés chez un modèle animal porteur d'une rétinopathie pigmentaire, à un stade où l'ensemble des bâtonnets a dégénéré, et où les cônes n'en sont qu'au début du processus de dégénérescence. Cette intervention ralentit de moitié la dégénérescence des cônes. Les chercheurs ont entrepris de caractériser les facteurs de survie des cônes, ce qui les a conduit à identifier la protéine Rod-derived Cone Viability Factor (RdCVF), sécrétée et exprimée spécifiquement par les bâtonnets, et qui disparaît après la première phase de dégénérescence des bâtonnets. Le gène assurant la synthèse de RdCVF, jusqu'ici inconnu (Txnl6), est essentiel à la survie des cônes chez la souris modèle. Cette avancée résulte d'une stratégie originale de génomique fonctionnelle menée depuis plus de 6 ans comprenant le criblage de 200 000 protéines. Les chercheurs vont maintenant s'attacher à élucider le mécanisme d'action de cette protéine et ils ont engagé des recherches précliniques pour son utilisation dans la thérapie des dégénérescences rétiniennes. Dans un deuxième temps, l'utilisation thérapeutique potentielle dans la DMLA sera explorée.
Cette découverte marque donc une étape essentielle vers la thérapeutique des dégénérescences rétiniennes et ouvre de nombreuses perspectives. En effet, les applications, basées sur l'administration de RdCVF, sont potentiellement larges puisque l'expression de la protéine est indépendante de l'anomalie génétique de la maladie. Elles sont mêmes envisageables à des stades avancés de la maladie (seulement 5% de cônes résiduels sont suffisants pour conserver une vision ambulatoire et la fonction de 50% des cônes assure une acuité visuelle normale).
Identification and characterization of rod-derived cone viability factor Thierry Léveillard (1), Saddeck Mohand-Saïd (1), Olivier Lorentz (1), David Hicks (1), Anne-Claire Fintz (1), Emmanuelle Clérin (1), Manuel Simonutti (1), Valérie Forster (1), Nükhet Cavusoglu (1), Frédéric Chalmel (2), Pascal Dollé (2), Olivier Poch (2), George Lambrou (3) & José-Alain Sahel (1,4)
(1) = Unité Inserm 592 "Laboratoire de Physiopathologie Cellulaire et Moléculaire et de la Rétine", hôpital St-Antoine, Paris
(2)= Institut de Génétique et de Biologie Moléculaire, Illkirch
(3) = Novartis pharma AG, ophthalmology research, Bâle, Suisse
(4)= Institute of ophthalmology, University College of London, Royaume-Uni
Nature Genetics, vol.36, n°7, pp 1-5, juillet 2004
15) Bibliographie
Ali RR, Sarra GM, Stephens C, et al. Restoration of Photoreceptor Ultrastructure and Function in Retinal Degeneration Slow Mice by Gene Therapy. Nat Genet 2000 Jul: 25(3):306-10.
Bareil C, Hamel C, Pallares-Ruiz N, Arnaud B, Demaille J, Claustres M. Molecular analysis of the rhodopsin gene in southern France : identification of the first duplication responsible for retinitis pigmentosa, c.998^999ins4. Ophtalmic Genet 1999, 20:173-82.
Berson EL. Retinitis pigmentosa: unfolding its mystery. Berman-Gund Laboratory for the Study of Retinal Degenerations, Harvard Medical School, MA 02114, USA. Proc Natl Acad Sci U S A 1996 May 14;93(10):4526-8.
Berson,E.L., Grimsby J.L., Adams,S.M., McGee T.L., Swekl E., Pierce,E.A., Sandberg,M.A., and Dryja,T.P. Clinical features and mutations in patients with dominant retinitis pigmentosa-1 (RP1). Invest.Ophthalmol.Vis.Sci. 2001, 42,2217-2224.
Bharadwaj,A.K., Kasztejna,J.P., Huq,S., Berson,E.L., and Dryja,T.P. (2000) Evaluation of the myosin VIIA gene and visual function in patients with Usher syndrome type I. Exp.Eye Res., 71,173-181.
Bok D, Yasumura D, Matthes MT, Ruiz A, Duncan JL, Chappelow AV, Zolutukhin S, Hauswirth W and LaVail MM. Effects of adeno-associated virus-vectored ciliary neurotrophic factor on retinal structure and function in mice with a P216L rds/peripherin mutation. Exp Eye Res 2002 74: 719-735.
Cayouette M, SB Smith, SP Becerra, and C Gravel (1999) Pigment Epithelium-Derived Factor delays the death of photoreceptors in mouse models of inherited retinal degenerations. Neurobiol Dis 6 : 523-532.
DeAngelis MM, Grimsby JL, Sandberg MA, Berson EL, Dryja TP. Novel mutations in the NRL gene and associated clinical findings in patients with dominant retinitis pigmentosa. Massachusetts Eye and Ear Infirmary, 243 Charles St, Boston, MA 02114, USA. Arch Ophthalmol 2002 Mar;120(3):369-75.
Delettre C, Lenears G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorges J, Turc-Carel C, Perret E, Astarie-Dequecker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP. Nuclear gene OPA1 encoding a mitochondrial dynamin-related protein is mutated in dominant optic atrophy. Nature Genetics 2000, 26:207-210.
Dryja TP. Molecular genetics of Oguchi disease, fundus albipunctatus, and other forms of stationary night blindness: LVII Edward Jackson Memorial Lecture. Department of Ophthalmology, Harvard Medical School and the Massachusetts Eye and Ear Infirmary, Boston, Massachusetts, USA. Am J Ophthalmol 2000 Nov;130(5):547-63.
Dryja,T.P., Hahn,L.B., Cowley,G.S., McGee,T.L., and Berson,E.L. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc.Natl.Acad.Sci.USA, 1991 88,9370-9374.
Hamel CP, Griffoin J-M, Bazalgette C, Lasquellec L, Duval P-A, Bareil C, Beaufrère L, Bonnet M, Eliaou C, Marlhens F, Schmitt-Bernard CF, Tuffery S, Claustres M, Arnaud B (2000). Génétique moléculaire des rétinopathies pigmentaires : identification de mutations de gènes CHM, RDS, RHO, RPE65, USH2A et XLRS1. J F Ophtalmol 23, 985-995.
Kedzierski, W., Bok D. and Travis, G.H. Non cell-autonomous photoreceptor degeneration in rds mutant mice mosaic for expression of a rescue transgene. J. Neurosci 1998 18: 4076-4082.
Mondal, MS, Ruiz A, Bok D and Rando RR Lecithin retinol acyltransferse contains cysteine residues essential for catalysis. Biochemistry 2000 39: 5215-5220.
Morimura,H., Saindelle-Ribeaudeau,F., Berson,E.L., and Dryja,T.P. Mutations in RGR, encoding a light-sensitive opsin homologue, in patients with retinitis pigmentosa. Nature Genet. 1999, 23,393-394.
Owens SL, Fitzke FW, Inglehearn CF, Jay M, Keen TJ, Arden GB, Bhattacharya SS, Bird AC. Ocular manifestations in autosomal dominant retinitis pigmentosa with a Lys-296-Glu rhodopsin mutation at the retinal binding site. Moorfields Eye Hospital, London. Br J Ophthalmol 1994 May;78(5):353-8.
Redmond TM, HAMEL CP. Genetic analysis of Rpe 65. From human disease to mouse model. Methods Enzymol 2000, 316:705-24.
Rosenfeld PJ, Hahn LB, Sandberg MA, Dryja TP, Berson EL.Low incidence of retinitis pigmentosa among heterozygous carriers of a specific rhodopsin splice site mutation. Berman-Gund Laboratory for the Study of Retinal Degenerations, Boston, Massachusetts, USA.
Rosenfeld,P.J., Cowley,G.S., McGee,T.L., Sandberg,M.A., Berson,E.L., and Dryja,T.P. A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nature Genet. 1992, 1,209-213.
Ruiz A, Winston A, Lim Y-H, Gilbert BA, Rando R, and Bok D. Molecular and biochemical characterization of lecithin retinol acyltransferase. J Biol Chem 1999 274: 3834-3841.
Ruiz A, Kuehn MH, Andorf J, Stone E, Hageman GS and Bok D. Organization of the lecithin retinol acyltransferase gene and mutation screening in various human hereditary retinal degenerations. Invest. Ophthalmol. Vis. Sci. 2001 42: 31-37.
Sarra G-M, Stephens C, de Alwis M, Bainbridge JWB, Smith AJ, Thrasher AJ, Ali RR (2001). Gene replacement therapy in the retinal degeneration slow (rds) mouse: the effect on retinal degeneration following partial transduction of the retina. Hum Molec Genet 10, 2353-2361.
Sharon,D., Bruns,G.A.P., McGee,T.L., Sandberg,M.A., Berson,E.L., and Dryja,T.P. X-linked retinitis pigmentosa: mutation spectrum of the RPGR and RP2 genes and correlation with visual function. Invest.Ophthalmol.Vis.Sci. 2000, 41,2712-2721.
Sippel KC, DeStefano JD, Berson EL, Dryja TP. Evaluation of the human arrestin gene in patients with retinitis pigmentosa and stationary night blindness. Ocular Molecular Genetics Institute, Harvard Medical School, Massachusetts Eye and Ear Infirmary, Boston, USA. Invest Ophthalmol Vis Sci 1998 Mar;39(3):665-70.
Vaithinathan,R., Berson,E.L., and Dryja,T.P. Further screening of the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. Genomics 1994, 21,461-463.
Wang M, Lam TT, Tso MOM, Naash MI (1997). Expression of a mutant opsin gene increases the susceptibility of the retina to light damage. Vis Neurosci 14, 55-62.
Wrigley, J.D.J., Ahmed, T., Nevett, C.L. & Findlay, J.B.C. (2000) Peripherin/rds influences membrane vesicle morphology. J Biol Chem , 275, 13191-13194.
Wrigley, J.D.J., Nevett, C.L. & Findlay, J.B.C. (2002) Topological analysis of peripherin/rds and abnormal glycosylation of the pathogenic P216L mutation.